
4. Sequenziamento di Acidi Nucleici
In questo capitolo analizzeremo una procedura sperimentale messa a punto da Fredrick Sanger nel 1977, con la quale è possibile leggere la sequenza lineare delle basi di un frammento di DNA. Il metodo viene oggi utilizzato anche da tutti i laboratori coinvolti nel Progetto Genoma Umano, che si pongono l'obiettivo di identificare tutti i geni presenti nel patrimonio genetico della nostra specie.
4.1 Blotting di DNA, RNA e proteine
Un frammento di restrizione contenente una specifica sequenza di basi può essere identificato ibridandolo con un filamento marcato di DNA complementare.
Una miscela di frammenti di restrizione viene separata tramite elettroforesi su gel di agarosio, quindi si denatura il DNA portandolo nella forma a singola catena e lo si trasferisce su un foglio di nitrocellulosa per capillarità (“blotting”) come descritto nella figura seguente:
Figura 4.1: Southern blotting. Identificazione di una sequenza specifica di DNA.
I vari frammenti di DNA mantengono sulla nitrocellulosa le stesse posizioni che avevano nel gel. Le posizioni possono essere determinate esponendo il foglio a un filamento di DNA a singola elica di sequenza nota (detta “probe” o sonda) marcato radioattivamente.
La sonda si lega con un filamento complementare e l’autoradiografia rivela quindi la posizione dell’insieme ibridato (cioè del complesso frammento di restrizione-sonda).
In questo modo si può identificare un frammento particolare in mezzo a milioni di altri frammenti.
Questa tecnica molto efficace prende il nome di Southern blotting in quanto ideata da Edwin Southern. La stessa procedura di analisi può essere seguita per individuare specifiche sequenze di RNA. La tecnica di analisi dell’RNA è stata chiamata Northern blotting.
Infine la Western blotting è una tecnica usata per rivelare una particolare proteina colorandola con uno specifico anticorpo. Southern, Northern e Western blotting sono noti anche come blotting di DNA, RNA e proteine.
4.2 Metodo dei didesossi di Sanger
Il DNA può essere sequenziato generando frammenti di DNA la cui lunghezza dipende dall’ultima base della sequenza. L’insieme di tali frammenti può essere generato attraverso una interruzione controllata della replicazione enzimatica. Questo metodo è stato sviluppato da Fredrick Sanger e dai suoi collaboratori.
Si realizza la stessa procedura, contemporaneamente, in quattro miscele di reazione distinte (A, G, T e C). In ciascuna di esse, si usa l’enzima DNA polimerasi per realizzare il complemento di una particolare sequenza di una molecola di DNA a singolo filamento.
La sintesi è cominciata da un frammento (detto primer), solitamente ottenuto tramite metodi chimici di sintesi, che è complementare alla parte di sequenza nota da altri studi.
Ciascuna miscela contiene, oltre ai quattro desossiribonucleotidi trifosfato (dATP, dTTP, dCTP, dGTP) marcati radioattivamente, anche un piccolo quantitativo di un analogo 2´-3´-didesossi (ddNTP).
In ognuna delle quattro miscele si ha un differente ddNTP: per esempio nella miscela A, ddATP (fig. 4.2), in quella G, ddGTP…
Figura 4.2 : strategia del metodo di Sanger nella miscela contenente ddATP (miscela A).
L’incorporazione di
questo analogo blocca la crescita ulteriore della catena. La concentrazione del ddNTP in ciascuna miscela è abbastanza
bassa in modo che la terminazione della catena possa avvenire solo
occasionalmente. La DNA polimerasi inserirà talvolta il nucleotide giusto e altre
volte il ddNTP analogo, bloccando la reazione e quindi la crescita della catena.
Nella miscela A di figura 5, la crescita della catena è interrotta ogni volta che viene inserito un ddATP; saranno quindi prodotti frammenti di varie lunghezze, tutti terminanti con ddATP.
Si osserva che il didesossi analogo di dATP è inserito solo dove
Figura 4.3: autoradiogramma del sequenziamento manuale di Sanger.

Questo tipo di sequenziamento è “manuale” in quanto la lettura
dell’autoradiogramma è affidata a un tecnico. Il metodo di Sanger è adatto per essere automatizzato, passando a
una rivelazione per fluorescenza.
È infatti possibile marcare il primer con un composto
fluorescente diversamente colorato per ciascuna delle quattro miscele di
reazione di terminazione ciascun primer quindi emette fluorescenza a lunghezza
d’onda diversa ed è facilmente individuabile.
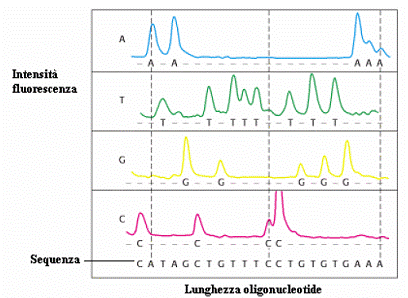
Eseguendo l’analisi esattamente come nel caso di sequenziamento manuale con quattro “corsie” per l’elettroforesi (e quindi quattro miscele distinte), l’indagine mediante fluorescenza produce un risultato analogo al caso precedente in cui però le bande separate di DNA sono rivelate dalla loro fluorescenza come essa emerge dal gel; la sequenza dei loro colori porta direttamente alla sequenza delle basi (fig. 4.4).
Figura 4.4: indagine mediante fluorescenza di frammenti oligonucleotidici
prodotti con il metodo dei dideossi.
In questo modo possono essere determinate sequenze lunghe fino a 500 basi. In alternativa, è possibile marcare i ddNTP, ciascuno con una differente etichetta fluorescente. Quando viene usato questo metodo, tutte e quattro le miscele del caso precedente possono coesistere in un’unica miscela e quindi è sufficiente una sola corsia su cui effettuare l’elettroforesi, evitando il rischio di variabilità elettroforetica fra una corsia e l’altra (fig. 4.5).
Figura 4.5: indagine mediante fluorescenza di frammenti
oligonucleotidici prodotti con il metodo dei dideossi.

La rivelazione fluorescente
è migliore perché elimina l’uso di reagenti radioattivi.
La marcatura più usata
attualmente è quella basata sui terminatori di catena chiamati “big-dyes derminator”, che consistono di didesossinucleotidi marcati con molecole con
sistema di trasferimento di energia da un donatore a un
accettore .
L’introduzione della marcatura fluorescente permette di passare dal sequenziamento manuale a
quello automatico che prevede una corsa elettroforetica su gel di poliacrilamide, su supporto a lastra o a capillare.
In entrambi i casi la migrazione dei vari frammenti è seguita rilevando le emissioni in fluorescenza a diverse lunghezze d’onda dei diversi fluorocromi dopo l’eccitazione provocata dal laser.
Le emissioni vengono raccolte e analizzate da una camera CCD (charge coupled device) che
elabora i diversi segnali di fluorescenza con elevata sensibilità.
La sequenza delle bande di DNA marcato viene visualizzata in un unico grafico detto elettroferogramma, caratterizzato da una successione di picchi di quattro colori diversi, che corrispondono alle emissioni fluorescenti dei diversi fluorocromi, ogni volta che i vari frammenti di diversa lunghezza nucleotidica raggiungono, lungo la corsa elettroforetica, la posizione del rilevatore (fig. 4.6).
Figura 4.6: esempio di elettroferogramma.
Rispetto al caso precedente sono associati colori differenti alle basi. In questo caso ad
A è associato il colore verde, a C il blu, a G il nero (trasformazione software del giallo)
e a T il rosso.

Come precedentemente visto, l’introduzione dell’elettroforesi capillare per la separazione dei frammenti marcati ha consentito un notevole aumento della processività. Sono stati inoltre sviluppati modelli di sequenziatori automatici che sono in grado di eseguire corse elettroforetiche multiple su apparecchi multicapillari.